7月14日,美國食品與藥品管理局(FDA)發(fā)布了藥物和診斷共同開發(fā)需遵循準則的指南草案,旨在將該指南作為實用的參考框架,幫助開發(fā)者推進治療產(chǎn)品和伴隨診斷的開發(fā),并幫助FDA工作人員審查這些產(chǎn)品。
FDA在指南草案中寫到,“IVD伴隨診斷和治療產(chǎn)品的共同開發(fā)對于精準醫(yī)療的推進至關(guān)重要。FDA試圖通過向開發(fā)者提供一套準則,幫助他們進行有效的共同開發(fā)以及執(zhí)行FDA的監(jiān)管要求,進而加速精準醫(yī)療方面的創(chuàng)新。”
Rx/Dx共同開發(fā)方案的監(jiān)管
對FDA來說,理想的Rx/Dx共同開發(fā)方案是,在藥物研發(fā)的最早期確定需要開發(fā)的伴隨診斷,兩者同時開發(fā)并同時進入市場。
兩年前FDA發(fā)布伴隨診斷的指南時闡述了上述理想方案。但后來FDA意識到,對藥物和伴隨診斷同時進行評估并不總是可行,因此允許將伴隨診斷的評估時間推后,先確定藥物是否對某種無藥可用的威脅生命的疾病有效。
通常情況下,F(xiàn)DA認為伴隨診斷屬于ClassIII、高風險產(chǎn)品,需要售前批準。但在共同開發(fā)指南中,F(xiàn)DA認為,一些Rx/Dx共同開發(fā)產(chǎn)品中的伴隨診斷可歸類為ClassII、中度風險產(chǎn)品,得到510(k)許可或de novo request后即可進入市場。
藥物和檢測開發(fā)的推進過程是明顯不同的,該指南提供了一個圖表,告訴開發(fā)者如何調(diào)整計劃以及何時應(yīng)該向FDA尋求意見。FDA建議,開發(fā)者需對治療和診斷的研發(fā)都有一定的了解,并且治療和診斷研發(fā)雙方都要出席與FDA藥物和診斷部門的會議。

FDA對伴隨診斷的定義是,一種必需的驗證藥物安全性和有效性的檢測。因此,F(xiàn)DA建議,伴隨診斷(CDx)的性能分析需在它應(yīng)用在藥物的臨床試驗前進行。當沒有足夠的數(shù)據(jù)證明一種試驗性新藥對患者的風險時,F(xiàn)DA會對這項研究下達“clinical hold(臨床試驗暫停)”通知。但FDA表示,伴隨診斷性能分析的不確定性不會導(dǎo)致這種暫停。
如果一種藥物的伴隨診斷之前未得到FDA批準用于這種特定用途,開發(fā)者必須提出申請并獲得器械臨床研究豁免(IDE)。在該指南草案中,F(xiàn)DA概述了IDE申請應(yīng)該包含的信息類型,以及在什么情況下體外診斷(IVD)可以不需要這個過程。該指南還討論了檢測開發(fā)者在利用訓(xùn)練樣本集、檢測設(shè)計變更的影響和IVD橋接試驗中應(yīng)該考慮的一些因素。
樣本采集
近一段時間,F(xiàn)DA一直在告誡開發(fā)者,醫(yī)生越來越多地預(yù)篩選病人,以明確他們進行生物標志物臨床試驗的資格,而這種行為會給Rx/Dx共同開發(fā)帶來問題。FDA表示,“預(yù)篩查會產(chǎn)生具有偏好性的臨床試驗人群,這個群體不能代表真實世界中使用IVD伴隨診斷的群體。因此,F(xiàn)DA強烈地反對挑選測試對象。”
FDA建議,開發(fā)者應(yīng)該要求臨床試驗參與單位提交的樣本來自于所有潛在候選檢測人群,而不是經(jīng)過本地檢測篩選出來的人群。FDA說,這樣才能夠評估IVD真實的分析能力,并且能夠確保意向治療人群不具有任何偏好性。
該指南草案還表示,F(xiàn)DA將在很多方面更加靈活,例如,允許在早期研究中使用臨床試驗分析,可展示NGS檢測中有代表性的標記物的分析驗證結(jié)果,當真實的病人樣本無法獲取時可利用人為樣本研究某種特定標記物。
但是FDA在指南中還指出,樣本采集對一個共同開發(fā)方案的成功至關(guān)重要,鼓勵開發(fā)者從所有招募的檢測對象中獲取樣本。這樣可確保臨床試驗分析進行不下去的情況下,開發(fā)者仍然能夠?qū)Π殡S檢測進行驗證且使其商業(yè)化。
生物標記物相關(guān)試驗的設(shè)計
該指南指出,制藥公司可以進行不同類型的生物標記物試驗設(shè)計,一種設(shè)計是按照陽性和陰性生物標記物狀態(tài)將患者隨機分為兩組,另一種設(shè)計是僅將陽性生物標記物狀態(tài)的患者隨機分到治療組。
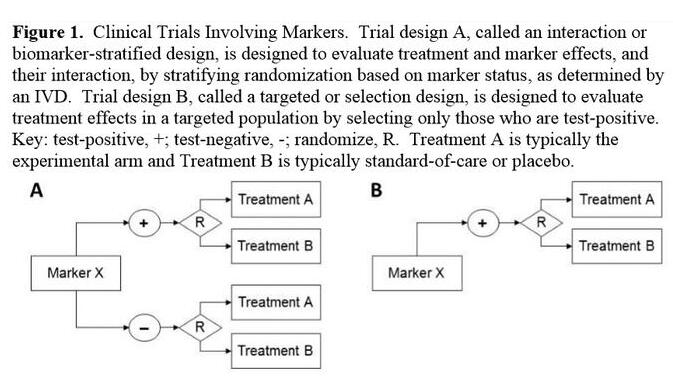
FDA表示,在衡量生物標記物的預(yù)后和預(yù)測價值方面,第一種試驗設(shè)計最有用。但是當有證據(jù)表明生物標記物陰性的患者對治療沒有應(yīng)答時,F(xiàn)DA會加速對只招募陽性生物標記物患者的精準藥物研究的審批工作。FDA還討論了一些方案,使制藥公司在一項前瞻性研究完成后,可以基于生物標記物回顧性地評估病人的應(yīng)答情況。
根據(jù)伴隨診斷是如何在藥物試驗中使用的,它將獲得一份聲明文件表明它的作用,是用于預(yù)測療效,監(jiān)測藥物劑量或者停藥,還是用于篩選進行臨床試驗的病人。FDA表示,“就篩選病人的伴隨診斷聲明來說,如果主要藥效試驗表明,該藥品對IVD篩選的人群具有足夠的安全性和有效性,這就說明該IVD得到了臨床驗證,其選擇的群體能從該治療產(chǎn)品中獲益。”
指南草案的意見征集
1998年,F(xiàn)DA批準了第一個乳腺癌治療藥物Herceptin(trastuzumab)和其伴隨診斷HercepTest。近年來,Rx/Dx共同開發(fā)產(chǎn)品快速增多,尤其是腫瘤類產(chǎn)品。該指南草案中關(guān)于共同開發(fā)的建議都來自FDA多年來批準這類產(chǎn)品的經(jīng)驗。
個性化醫(yī)學(xué)聯(lián)盟(Personalized Medicine Coalition,PMC)的執(zhí)行副總裁Amy Miller說,“FDA藥品和診斷部門開發(fā)了內(nèi)部流程,使腫瘤藥物通過FDA審查。我們建議FDA利用這些經(jīng)驗,為開發(fā)者和FDA其他審查領(lǐng)域的員工起草一份“如何做”的指南,所以他們就制定了這份指南草案。”
該共同開發(fā)指南草案已經(jīng)制定了很長一段時間了,多年來FDA與利益相關(guān)人士在研討會和行業(yè)會議上就文件中的相關(guān)準則進行了交流。Miller說,PMC會進一步研究這份草案并提出改進意見。公眾有90天時間對該指南草案提出意見。








 相關(guān)新聞
相關(guān)新聞










