



業(yè)務(wù)咨詢
中國:
Email: marketing@medicilon.com.cn
業(yè)務(wù)咨詢專線:400-780-8018
(僅限服務(wù)咨詢,其他事宜請撥打川沙總部電話)
川沙總部電話: +86 (21) 5859-1500
海外:
+1(781)535-1428(U.S.)
0044 7790 816 954 (Europe)
Email:marketing@medicilon.com




藥物研發(fā)的主要思路是通過藥物來抑制或激發(fā)疾病發(fā)病機(jī)制中的某一個(gè)靶點(diǎn)。但是,近年來系統(tǒng)生物學(xué)為不斷發(fā)展的藥物研發(fā)提供了一種新的思路:多靶點(diǎn)藥物治療。同時(shí),對疾病發(fā)病機(jī)制的進(jìn)一步研究和認(rèn)識(shí)也表明了,單獨(dú)靶點(diǎn)的抑制或激發(fā),在復(fù)雜疾病治療中有局限性。多靶點(diǎn)藥物治療,可使藥物同時(shí)作用于多個(gè)靶點(diǎn),對各靶點(diǎn)的作用可以產(chǎn)生協(xié)同效應(yīng),使總效應(yīng)大于各單效應(yīng)之和,達(dá)到最佳的治療效果。
多靶點(diǎn)藥物的概括
多靶點(diǎn)藥物按組分的不同可以分為三種形式。其一為:多種藥物聯(lián)合用藥,缺點(diǎn)是所含藥物彼此間容易發(fā)生相互作用而產(chǎn)生不良反應(yīng)。其二為:多組分單藥片,即在一個(gè)給藥單位中含有多種活性組分。其三為:某一單組分藥物可以同時(shí)選擇性作用于多個(gè)分子靶點(diǎn),即嚴(yán)格意義上的多靶點(diǎn)藥物。單組分藥物在藥物代謝上是較聯(lián)合用藥和多組分的藥物有更多優(yōu)勢的,因其可以克服各組分相互作用產(chǎn)生的不良反應(yīng)。
由于單靶點(diǎn)藥物只能調(diào)控疾病過程中的一個(gè)點(diǎn),而各種臨床重大疾病通常是多重因素導(dǎo)致的結(jié)果,且其病理機(jī)制及疾病治療過程非常復(fù)雜。因此,單靶點(diǎn)藥物往往會(huì)有治療效果不佳、藥物篩選效率不高等缺點(diǎn)。而多靶點(diǎn)藥物可以同時(shí)作用于多個(gè)病理環(huán)節(jié)、多種發(fā)病機(jī)制而產(chǎn)生協(xié)同作用效果,從而提高藥物的療效。
藥物靶標(biāo)通常處于多個(gè)信號(hào)通路中,具有多重生物學(xué)功能,過分激活或抑制某一生物靶標(biāo),在干預(yù)一種生物學(xué)功能的同時(shí),也可以影響其它正常生物學(xué)功能,從而導(dǎo)致毒副作用的發(fā)作。多靶點(diǎn)藥物可以更好的平衡多個(gè)病理因素間的關(guān)系,可以在相對更低的血藥濃度水平,產(chǎn)生單靶點(diǎn)藥物需要高濃度才能產(chǎn)生的生物學(xué)效應(yīng),且對生物靶標(biāo)一般具有弱親和力的特點(diǎn),因而可以減少藥物的不良反應(yīng)。
生物機(jī)體是一個(gè)復(fù)雜的可自我調(diào)節(jié)和平衡的系統(tǒng),長期使用某一單靶點(diǎn)藥物治療疾病,可以誘導(dǎo)機(jī)體內(nèi)部的適應(yīng)性變化從而激活對抗保護(hù)機(jī)制或者旁路代償機(jī)制等,使疾病對該種藥物不再敏感,造成藥物的耐藥性。而多靶點(diǎn)藥物可以通過同時(shí)干預(yù)疾病的主要致病靶標(biāo)及其代償信號(hào)通路或者其保護(hù)性信號(hào)通路而減少疾病對藥物產(chǎn)生的耐藥性。
多靶點(diǎn)藥物需要協(xié)調(diào)平衡多方面的參數(shù)使之處于適度區(qū)間,如藥效學(xué)方面需要考慮靶標(biāo)組合的合理性、活性的平衡性和靶標(biāo)的選擇性;藥動(dòng)學(xué)方面,要考慮ADME各方面特性的適當(dāng)與否;在化學(xué)方面,要考慮其理化性質(zhì)的合適與否等;靶標(biāo)組合方面,它要求選擇疾病病理機(jī)制中最關(guān)鍵的幾個(gè)靶標(biāo)進(jìn)行組合,并保證靶標(biāo)組合的合理性;活性平衡方面,多靶點(diǎn)藥物對各靶點(diǎn)的作用強(qiáng)度接近而不宜差別太大;靶標(biāo)選擇方面,多靶點(diǎn)藥物只選擇作用于所確定的靶標(biāo)組合,而不應(yīng)對其他靶標(biāo)有多余的活性,以減小不必要的副作用。
盡管存在很多問題和困難,隨著現(xiàn)代系統(tǒng)生物學(xué)、化學(xué)生物學(xué)以及計(jì)算機(jī)輔助藥物設(shè)計(jì)技術(shù)等的發(fā)展,多靶點(diǎn)藥物的研究也在一步步的拓展。
已上市多靶點(diǎn)藥物的概況
隨著多靶點(diǎn)藥物發(fā)現(xiàn)技術(shù)的不斷成熟,已經(jīng)有越來越多的多靶點(diǎn)藥物進(jìn)入臨床應(yīng)用,尤其是在癌癥、糖尿病、病毒和細(xì)菌感染等復(fù)雜性疾病中。近年來,F(xiàn)DA先后批準(zhǔn)了多個(gè)多靶點(diǎn)酪氨酸激酶抑制劑上市,包括2005年獲批的索拉非尼、2006年獲批的達(dá)沙替尼、2007年獲批的舒尼替尼和拉帕替尼。另外,多靶點(diǎn)藥物還包括心血管藥物奧馬曲拉、特波格雷、普齊地洛;中樞神經(jīng)系統(tǒng)藥物拉多替吉、奧氮平、卡巴拉汀等等。
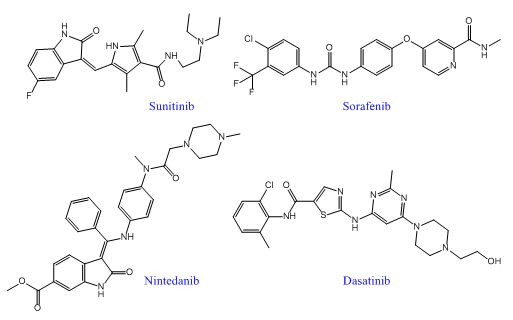
索拉菲尼是第一個(gè)用于腫瘤治療的多靶點(diǎn)激酶抑制劑,能抑制RAF-1、B-RAF的絲氨酸/蘇氨酸激酶活性,同時(shí)可以抑制VEGFR-2、VEGFR-3、PDGF-β、KTI、FLT-3等多種受體的酪氨酸激酶活性。索拉菲尼具有雙重抗腫瘤作用,既可以阻斷有由RAF/MEK/ERK介導(dǎo)的細(xì)胞信號(hào)轉(zhuǎn)導(dǎo)通路,直接抑制腫瘤細(xì)胞的增殖,還可以通過作用于VEGF、PDGF-β等受體,抑制新生血管的形成和阻斷腫瘤細(xì)胞的營養(yǎng)供應(yīng)和代謝而達(dá)到遏制腫瘤生長的目的。在2005年12月,美國FDA就批準(zhǔn)其用于治療晚期腎癌,后2007年11月,美國FDA再次批準(zhǔn)其用于無法切除治療的晚期肝癌。該藥在我國分別于2006年和2008年被批準(zhǔn)用于晚期腎癌和晚期肝癌的治療。2011~2015連續(xù)五年,索拉非尼的年銷售額超過10億美元。
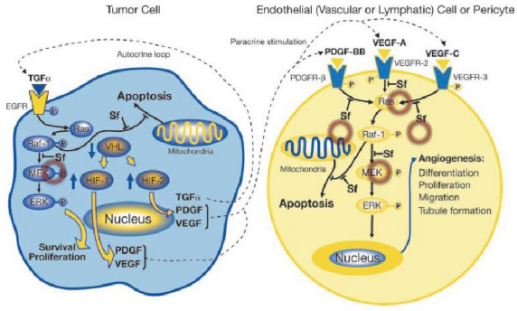
索拉非尼抗靶向細(xì)胞增殖和血管生成示意圖
舒尼替尼是一種新型吲哚酮類口服、選擇性多靶點(diǎn)酪氨酸激酶抑制劑,除抑制VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-α、PDGFR-β的活性外,同時(shí)也抑制幾種其他相關(guān)的酪氨酸激酶的活性,包括c-KIT、FLT-3和RET受體,具有抗血管生成和抗腫瘤活性的雙重作用。美國FDA已經(jīng)批準(zhǔn)其用于晚期腎細(xì)胞癌,胃腸間質(zhì)瘤和晚期胰腺神經(jīng)內(nèi)分泌瘤。臨床前期研究表明,舒尼替尼能夠有效抑制人NSCLC異種移植瘤模型的生長。多項(xiàng)臨床研究評估了舒尼替尼在晚期NSCLC治療中的作用,初步顯示其在多線治療后的晚期NSCLC中仍能取得一定的療效,能改善患者生存,且毒性耐受。
阿西替尼是新一代強(qiáng)效的多靶點(diǎn)藥物,其主要的作用靶點(diǎn)為VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-β和c-KIT,是目前對VEGFR信號(hào)通路抑制率最強(qiáng)的酪氨酸激酶抑制劑,在多種實(shí)體腫瘤中顯示出良好的抗腫瘤活性。FDA已批準(zhǔn)用于晚期腎細(xì)胞癌的二線治療。在一項(xiàng)阿西替尼單藥治療晚期NSCLC的II期臨床研究中,顯示了其在NSCLC患者中良好的抗腫瘤活性和安全性。
尼達(dá)尼布為三重酪氨酸激酶抑制劑,靶點(diǎn)包括VEGF、PDGF和FGF,也可抑制MAPK和AKT的激活。體外研究表明,尼達(dá)尼布能持續(xù)抑制VEGFR-2達(dá)30小時(shí)以上。I-II期臨床研究顯示,尼達(dá)尼布對第一、二線治療失敗的復(fù)發(fā)轉(zhuǎn)移性NSCLC及局部晚期NSCLC有效,部分患者表現(xiàn)為腫瘤縮小,病情穩(wěn)定。
新多靶點(diǎn)抑制劑的發(fā)現(xiàn)
抑制p53-MDM2蛋白結(jié)合和組蛋白去乙酰化酶(HDACs)是抗腫瘤藥物開發(fā)的重要靶點(diǎn)。受MDM2和HDACs協(xié)同作用的啟發(fā),Shipeng He等人發(fā)現(xiàn)了第一個(gè)MDM2/HDACs雙抑制劑(14d),對MDM2/HDACs這兩個(gè)靶點(diǎn)都有很好的活性,且其抗腫瘤機(jī)制在癌細(xì)胞中得到驗(yàn)證,為新型抗腫瘤藥物的開發(fā)提供了一種很有前途的小分子抑制劑[1]。

p53是一種轉(zhuǎn)錄因子,在預(yù)防腫瘤發(fā)展中扮演著重要角色。大約50%的人類癌癥與p53的失活有關(guān)[2]。MDM2基因上的rs2279744位點(diǎn)發(fā)生突變后,其蛋白產(chǎn)物能夠與 P53 蛋白結(jié)合并增強(qiáng)其降解,從而導(dǎo)致 P53 蛋白的抑癌作用減弱。因此,抑制p53-MDM2蛋白質(zhì)結(jié)合成為了一種新興有前途的癌癥治療策略[3]。研究發(fā)現(xiàn),單一化合物同時(shí)調(diào)制多個(gè)靶點(diǎn)可能會(huì)產(chǎn)生更加卓越的功效以及更少的副作用[4]。組蛋白去乙酰化酶(HDACs),一種表觀遺傳酶,在調(diào)節(jié)腫瘤抑制基因的表達(dá)方面發(fā)揮著至關(guān)重要的作用[5]。然而,大多數(shù)HDAC抑制劑需要與其他抗腫瘤藥物結(jié)合使用,實(shí)現(xiàn)協(xié)同效應(yīng)[6]。因此,,Shipeng He等人合理研究設(shè)計(jì)了首個(gè)MDM2/HDAC雙抑制劑(14d)。在A549異種移植模型中,雙抑制劑14d的抗腫瘤效果良好,證明了這種新型多靶向抗腫瘤藥物設(shè)計(jì)策略的價(jià)值。
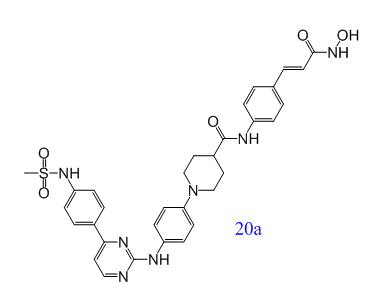
在臨床上,白血病患者常因化療療效有限,且易受侵襲性真菌病原感染。Yahui Huang等人提出了一種新的治療策略:小分子抑制劑可以同時(shí)治療白血病和侵襲性真菌感染(IFIs)。新型Janus激酶2(JAK2)和組蛋白去乙酰化酶(HDAC)雙重抑制劑對血液細(xì)胞系具有較強(qiáng)的抗增殖活性。其中,化合物20a是一種高度活性和選擇性的JAK2/ HDAC6雙重抑制劑,在幾種急性髓系白血病(AML)模型中顯示出極好的體內(nèi)抗腫瘤作用,并可與氟康唑協(xié)同治療耐藥的白色念珠菌感染[7]。
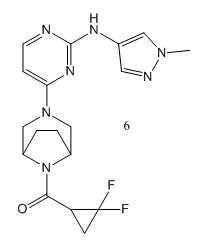
Christel J Menet提到小分子抑制劑6,它具有較好的選擇性和良好的口服生物利用度,并在JAK1和TYK2的雙重抑制給藥方案中取得了臨床益處。在最近的一篇論文中[8],研究結(jié)果表明,TYK2 / JAK1抑制劑6有在健康受試者和斑塊性銀屑病患者中有良好的安全性和耐受性,此結(jié)論支持抑制劑6在銀屑病及其他炎性疾病治療中值得進(jìn)一步臨床研究。
從目前藥物研發(fā)的情勢來看,創(chuàng)新藥物的研發(fā)是迫在眉睫的,而提高藥物研發(fā)的綜合水平是我國所面臨的主要難題。因此,在對已知靶點(diǎn)合理的、最大可能的利用是可實(shí)行且回報(bào)率高的研究方向,也應(yīng)多關(guān)注于多靶點(diǎn)藥物的設(shè)計(jì),從而獲得更多新的多靶點(diǎn)抑制劑,給疾病患者帶來更好的治療效果。
參考文獻(xiàn):
1,ShipengHe, Guoqiang Dong, Shanchao Wu, Kun Fang, Zhenyuan Miao, Wei Wang and ChunquanSheng.Small Molecules Simultaneously Inhibitingp53-Murine Double Minute 2 (MDM2) Interaction and Histone Deacetylases (HDACs):Discovery of Novel Multitargeting Antitumor Agents.J. Med. Chem. 61, 16, 7245-7260.
2,Hainaut,P. Hollstein, M. P53 and human cancer: the first ten thousand mutations. Adv.Cancer Res. 1999, 77, 81– 137.
3,Wang,S. Zhao, Y. Bernard, D. Aguilar, A. Kumar, S. Targeting the MDM2-p53protein-protein interaction for new cancer therapeutics. Top. Med. Chem. 2012,8, 57– 79.
4,Singh,A. K. Chauhan, S. S. Singh, S. K. Verma, V. V. Singh, A. Arya, R. K.Maheshwari, S. Akhtar, M. S. Sarkar, J. Rangnekar, V. M. Chauhan, P. M. S.Datta, D. Dual targeting of MDM2 with a novel small-molecule inhibitorovercomes TRAIL resistance in cancer. Carcinogenesis 2016, 37, 1027– 1040.
5,Ropero,S. Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol.Oncol. 2007, 1, 19– 25.
6,Ong,P. S. Wang, X. Q. Lin, H. S. Chan, S. Y. Ho, P. C. Synergistic effects ofsuberoylanilide hydroxamic acid combined with cispiatin causing cell cyclearrest independent apoptosis in platinum-resistant ovarian cancer cells. Int.J. Oncol. 2012, 40, 1705– 1713.
7,YahuiHuang, Guoqiang Dong, Huanqiu Li, Na Liu, Wannian Zhang and Chunquan Sheng.Discovery of Janus Kinase 2 (JAK2) andHistone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for theCombinational Treatment of Leukemia and Invasive Fungal Infections.J. Med. Chem. 2018, 61, 6056?6074.
8,Banfield,C. Scaramozza, M. Zhang, W. Kieras, E. Page, K. M. Fensome, A. Vincent, M.Dowty, M. E. Goteti, K. Winkle, P. J. Peeva, E. The Safety, Tolerability,Pharmacokinetics, and Pharmacodynamics of a TYK2/JAK1 Inhibitor (PF-06700841)in Healthy Subjects and Patients With Plaque Psoriasis. J. Clin. Pharmacol.2018, 58 (4), 434–447.
 相關(guān)新聞
相關(guān)新聞川沙總部
地址: 上海市浦東新區(qū)川大路585號(hào)
郵編: 201299
電話: +86 (21) 5859-1500(總機(jī))
傳真: +86 (21) 5859-6369
業(yè)務(wù)咨詢
中國:
Email: marketing@medicilon.com
業(yè)務(wù)咨詢專線:400-780-8018
(僅限服務(wù)咨詢,其他事宜請撥打川沙
總部電話)
海外:
Email:?marketing@medicilon.com
Tel: +1 (617) 888-9294(U.S.)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)











